„”
We discovered the CLC gene family in 1990 by expression cloning of the voltage-gated Cl- channel from the electric organ of the marine ray Torpedo [1]. This channel, which we baptized ClC-0, is the founding member of the CLC gene family which is present from bacteria to men. In humans, it comprises 9 members (Figure 1) (for review, see [2]). Originally thought to only represent plasma membrane Cl- channels, many of these proteins rather function as electrogenic Cl-/H+-exchangers in intracellular membranes. Mutations of CLC genes underlie several human genetic diseases. We have shown that some CLC proteins associate with other, structurally unrelated proteins, mutations in which cause pathologies that overlap with those observed with the loss of their respective CLC partners.
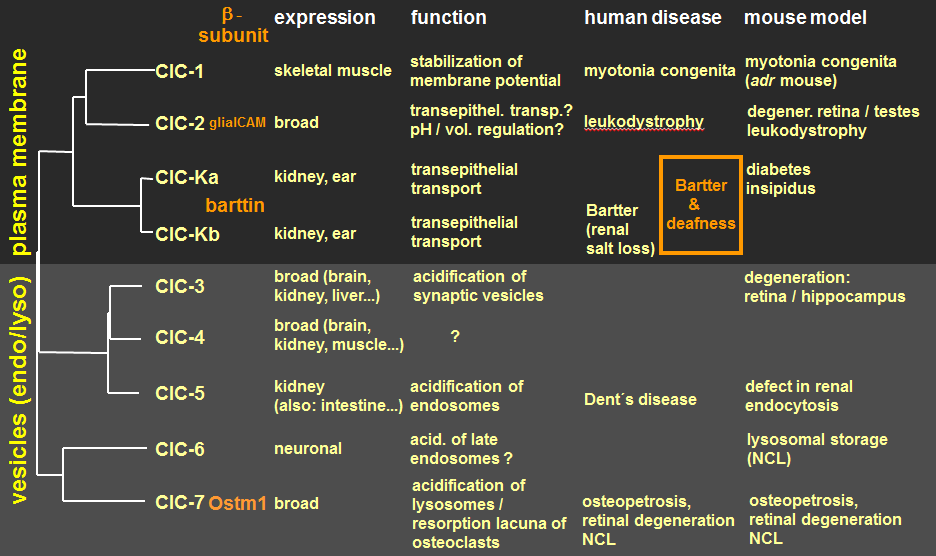
Figure 1. The CLC gene family of Cl- channels and Cl-/H+-exchangers. Associated structurally unrelated β-subunits include the cell adhesion molecule GlialCAM (mutations in which underlie a distinct form of leukodystrophy) for ClC-2, the 2-membrane span membrane protein barttin (mutations in which lead to Bartter syndrome with deafness) for both ClC-K isoforms, and for ClC-7 Ostm1, with disruption of either gene leading to osteopetrosis associated with neurodegeneration.
Important new insights were obtained from our disruption of CLC genes in mice. For instance, the disruption of the chloride transporter ClC-7 unexpectedly led to osteopetrosis [3], a hypercalcification of bone. This phenotype is due to a dysfunction of osteoclasts, the cells that are responsible for bone degradation. ClC-7 currents may balance the electrogenic transport of the H+-ATPase that acidifies the resorption lacuna (Figure 2).
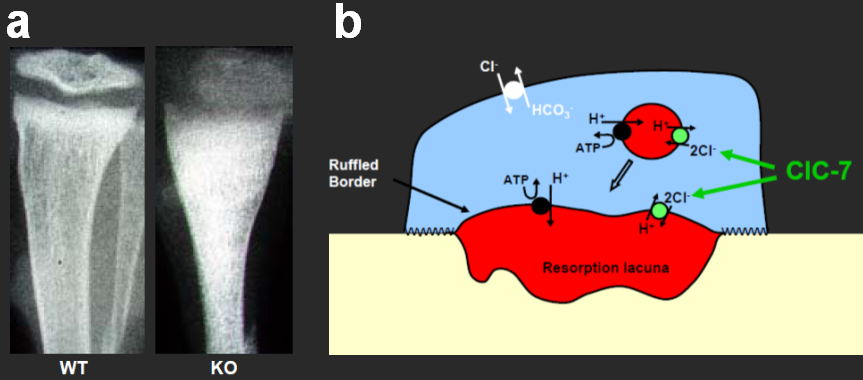
Figure 2. Role of ClC-7 in osteopetrosis. a, X-ray of bones from wild-type (WT) and ClC-7 KO mice reveals hypercalcification in the KO. b, model for the acidification of the osteoclast resorption lacuna.
Motivated by these findings, we identified mutations in either the ClC-7 chloride transporter [3], or in the a3 subunit of the H+-ATPase [4], in human patients with severe juvenile osteopetrosis. ClC-7 is normally present in a lysosomal compartment, but is inserted into the plasma membrane of osteoclasts. Similarly, our results show that ClC-3 through ClC-6 reside normally in membranes of intracellular compartments, where they may contribute to their acidification by electrical shunting of the proton ATPase. ClC-5, a Cl-/H+ exchanger mutated in the kidney stone disorder Dent’s disease [5], is present in renal endosomes [6]. Our ClC-5 knock-out mouse model [7] revealed that an impaired endosomal acidification led to a broad defect in proximal tubular endocytosis. This entails secondary changes in calciotropic hormones, eventually resulting in the hyperphosphaturia and hypercalciuria (and kidney stones) in Dent’s disease. The related ClC-3 putative Cl-/H+ exchanger is also present in endosomes, as well as in synaptic vesicles [8]. As the uptake of neurotransmitters into synaptic vesicles is coupled to the proton gradient, this may have interesting consequences for synaptic transmission. Surprisingly, the knock-out of ClC-3 led to nearly complete degeneration of the hippocampus [8].
In the classical model of vesicular acidification (Figure 3a), the electrical current of the vesicular H+-ATPase is neutralized by Cl- influx through a Cl- channel. The discovery that vesicular CLCs are 2Cl-/H+-exchangers rather than Cl- channels [9, 10] therefore came as a surprise because a role in acidification rather seemed counterintuitive.

Figure 3. Back to classics. We generated ClC-5 and ClC-7 knock-in mice in which we converted the WT 2Cl-/H+-exchangers ClC-5 and ClC-7 (b) with point mutations into mere Cl- conductances (c) to conform to the classical model (a) of vesicular acidification.
To elucidate whether vesicular CLCs just serve to neutralize proton pump currents, we converted ClC-5 and ClC-7 in mice to pure Cl- conductances [11, 12]. Any phenotype apparent in those mice cannot be ascribed to a defect in vesicular acidification. Surprisingly, these mice displayed grossly the same phenotype as the corresponding KO mice, suggesting an important role of vesicular Cl- accumulation. Another recent ClC-7 knock-in mouse [13] revealed that not only ion transport activity, but also (so far unknown) protein-protein interactions account for ClC-7 functions.
ClC-2 is a widely expressed plasma membrane Cl- channel that is slowly activated by inside-negative voltage or cell swelling [14, 15]. We showed previously that disruption of ClC-2 in mice leads to testicular and retinal degeneration [16], as well as leukodystrophy [17]. Recently van der Knaap and colleagues showed that human ClC-2 mutations also lead to leukodystrophy, a disease associated with vacuolization of the white matter (glia) of the central nervous system. Together with Raúl Estévez we have recently shown that ClC-2 associates with GlialCAM, an Ig-like cell adhesion cell surface molecule, and that this interaction profoundly changes ClC-2 gating [18]. Importantly, GlialCAM mutations also lead to human leukodystrophy, as do mutations in MLC1, a protein interaction partner of GlialCAM that displays several transmembrane domains. We recently generated GlialCAM mouse models and compared their CNS pathology to those of ClC-2 and MLC1 KO mice [19]. Unexpectedly, the localization, abundance and biophysical properties of ClC-2 depended on both GlialCAM and MLC1, suggesting changes in ClC-2 as a common pathogenic factor in the respective diseases.
The Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) is part of the Forschungsverbund Berlin e.V. (FVB), which legally represents seven non-university research institutes - members of the Leibniz Association - in Berlin.
Leibniz-Forschungsinstitut für Molekulare Pharmakologie im Forschungsverbund Berlin e.V. (FMP)
Campus Berlin-Buch
Robert-Roessle-Str. 10,
13125 Berlin, Germany