„”
The term '''lysosomal storage disease''' summarizes a number of genetically determined metabolic diseases that are due to incorrect or insufficient function of lysosomes. These cellular organelles are important both as “cellular waste disposal” and for the regulation of cellular metabolism. If lysosomal function is compromised, substances that normally would be degraded may accumulate in the affected cells. This may impair their function and may eventually lead to cell death. In the central nervous system, which is often affected because adult neurons are unable to regenerate, this can lead to neurodegeneration.
Researchers from the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and the Max-Delbrück-Centrum für Molekulare Medizin (MDC), in close collaboration with colleagues from Rome, Hamburg and the USA, have now found and characterized the gene defect underlying a novel severe form of neurodegenerative disease: A mutation in the CLCN6 gene in three unrelated children from Italy, Germany and the USA, leads to severe developmental delay, intellectual disability, hypotonia severely affecting muscle tone, respiratory insufficiency, visual impairment, and early-onset brain atrophy.
Ion transporter ClC-6 is a member of the chloride channel family
Human geneticists, including the study’s co-leader Marco Tartaglia from Rome and Kerstin Kutsche from Hamburg, independently discovered the same point mutation in their young patients and asked Prof. Thomas Jentsch and his team to examine possible effects of the mutation on the transport properties of ClC-6 and its cellular functions. Jentsch, the discoverer of the CLC chloride channel family, had already found or characterized different disease-causing mutations in almost all nine CLC genes. These are associated with a broad spectrum of different pathologies. Only the genes encoding the ion transporters ClC-3 and ClC-6 had not yet been found to be mutated in human disease. '''About fifteen years ago, we had generated a ClC-6 knockout mouse and found that it displayed mild neuronal lysosomal storage. However, our search for patients with similar loss function mutations in ClC-6 was unsuccessful,''' explains Prof. Jentsch. '''Now we have identified a different type of ClC-6 mutation in a much more severe human disease.”
The presence of exactly the same mutation in three independent patients displaying the same disease pattern already indicated a causal role of the mutation. But only the functional analysis in cell culture brought final certainty and led to the classification as lysosomal disease. '''Our cell cultures experiments clearly show that increased ion transport by the mutated ClC-6 affects lysosomes and thereby prove the deleterious effect of the mutation. Based on these results, and taking into account our previous mouse model, we assume that the novel disease can be classified as lysosomal storage disease,''' explains Thomas Jentsch. However, definitive proof of this classification would require post-mortem examination of brain slices from patients or a novel mouse model carrying the same mutation.
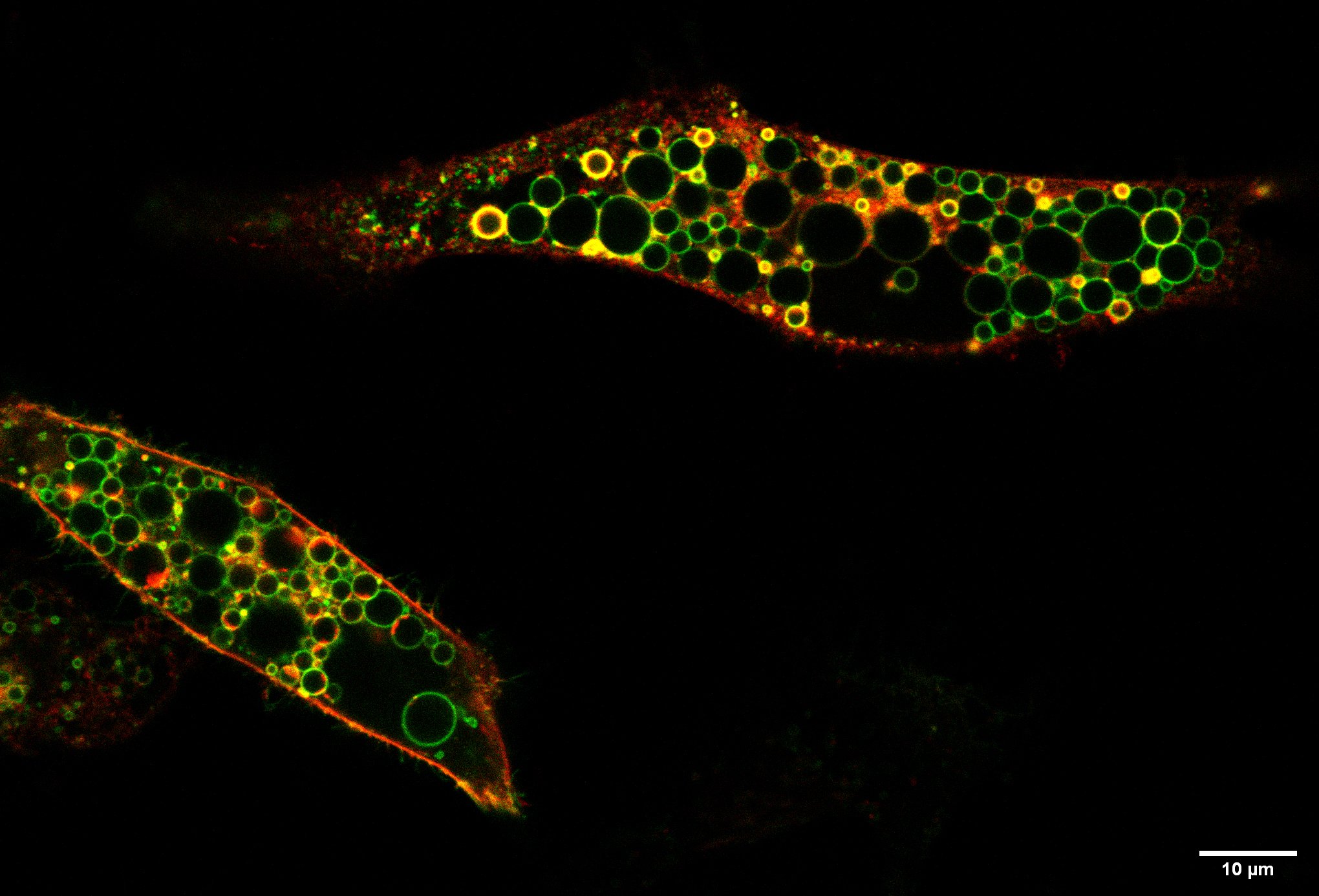
More chloride uptake leads to abnormally large, lysosome-like vesicles
Unlike the chloride channels ClC-1, -2, -3 and -K, the chloride/proton exchangers ClC-3, -4, -5, -6, and -7 are not located on the plasma membrane but in intracellular membranes, mainly on endosomes and lysosomes. In previous studies, Jentsch and coworkers identified mutations of ClC-7 as the cause of a form of lysosomal storage disease associated with osteopetrosis, and mutations of ClC-4 lead to intellectual deficits. While ClC-7 is found on lysosomes, ClC-6 is predominantly located on late endosomes, kind of lysosome precursors.
The Berlin team found that the patients’ mutation, in contrast to the loss of ClC-6 in their previous knock-out mouse model, caused a hyperactive ClC-6: The transport of chloride and protons was highly increased and was no longer modulated by pH. Normally acidic pH, as gradually achieved in the transition from endosomes to lysosomes, inhibits the transporter. This regulation is missing in the disease-causing mutant. The increased, unregulated ion transport - a pathological gain of function - resulted in drastically enlarged, lysosome-like vesicles in cells that were made to produce the mutated ClC-6. According to Jentsch, this pathological gain of function can explain the children's disease. '''Vesicles carrying the mutated ClC-6 in their membrane are pathologically enlarged by an increased uptake of chloride, which is later followed by water. This uptake is driven by the ClC-6-mediated exchange for protons which are abundantly present in the acidic interior of vesicles. This severely impairs the function of lysosomes and, in the long run, probably leads to lysosomal storage in neurons, cells that are unable to proliferate. The tissue distribution of ClC-6, which is found almost exclusively in neurons, contributes to the predominantly neurological disease '''.
'''The present work highlights the importance of ion transport for the endosomal-lysosomal pathway,''' says Jentsch. '''We see a broad spectrum of genetic diseases that are caused by mutations in vesicular CLCs or in different intracellular channels.” Very different organs can be affected: For example, mutations in the endosomal ClC-5 lead to kidney stones and protein loss into the urine, as Jentsch’s team showed a long time ago.
Jentsch is confident that also the ClC-3 exchanger will soon be linked to a genetic disease - a KO mouse previously published by the group shows dramatic neurodegeneration. Together with the current finding, this would link all nine CLCN genes to human genetic disease. '''The gap is closing''', says Jentsch, '''and we can see very clearly how important basic research - we had cloned the first CLC from an electric fish - is for the diagnosis and understanding of human disease'''.
Text: Beatrice Hamberger
Publication
Polovitskaya M., Barbini C., Martinelli D., Harms F., Cole S., Calligari P.,Bocchinfuso G., Stella L., Ciolfi A., Niceta M., Rizza T., Shinawi M., Sisco K., Johannsen J., Denecke J., Carrozzo R., Wegner D., Kutsche K., Tartaglia M., Jentsch T.J., A recurrent gain-of-function mutation in CLCN6, encoding the ClC-6 Cl-/H+-exchanger, causes early-onset neurodegeneration, American J. Human Genetics, 107; DOI: 10.1016/j.ajhg.2020.11.004
The Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) is part of the Forschungsverbund Berlin e.V. (FVB), which legally represents seven non-university research institutes - members of the Leibniz Association - in Berlin.
Leibniz-Forschungsinstitut für Molekulare Pharmakologie im Forschungsverbund Berlin e.V. (FMP)
Campus Berlin-Buch
Robert-Roessle-Str. 10,
13125 Berlin, Germany