„”
"subtitle"
CLC CHLORIDE CHANNELS AND TRANSPORTERS. Back in 1990, we achieved a major breakthrough by molecularly identifying the first voltage-gated Cl channel, which we isolated from the Torpedo electric organ and subsequently named ClC-0. It defined the CLC gene family, which in mammals comprises nine members. These proteins function either as plasma membrane Cl channels or as intracellular (vesicular) 2Cl/H exchangers. Our lab investigated all CLC proteins in considerable detail and discovered several important pathologies in mice and humans that are due to mutations in their genes. (Recent reviews in J Physiol and Physiol. Rev.)
In the CLC area, our research is currently focusing on the role of vesicular CLCs in endosomal/lysosomal function and ion homeostasis, and on ClC-2 in various pathologies, in particular its recently discovered role in primary hyperaldosteronism.
Vesicular CLC proteins may help in the luminal acidification of endosomes and lysosomes. In the classical picture of vesicular acidification, electrical currents of the H+-ATPase are neutralized by a Cl- channel (left). However, we have shown that endosomal CLC proteins, instead of being Cl- channels, are rather Cl-/H+ exchangers (centre), raising the question what this exchange is good for. In our most recent work, we generated knock-in mice in which we converted selected CLC exchangers into channels using single point mutations (right panel). These mice should display normal acidification of endosomes (for ClC-5) or lysosomes (for ClC-7). Surprisingly, both mouse models (Clcn5unc and Clcn7unc, unc for uncoupled from protons) display phenotypes (impaired endocytosis or neurodegeneration) that largely overlap with those of the respective KOs. We suggest that there is an important, previously unrecognized role of luminal chloride concentration (Weinert et al., Novarino et al.; Science 2010).

We generated ClC-5 and ClC-7 knock-in mice in which we converted the WT 2Cl-/H+-exchangers ClC-5 and ClC-7 (b) with point mutations into mere Cl- conductances (c) to conform to the classical model (a) of vesicular acidification.
More recently, we extended these findings to ClC-3 (Weinert et al., EMBO J 2020). Whereas disruption of ClC-3 leads to neurodegeneration (complete loss of the hippocampus), its conversion to a chloride conductor in Clcn3unc/unc mice surprisingly did not lead pathology. However, this can be explained by compensation by ClC-4, with which ClC-3 forms heterodimers. The loss of ClC-3, but not its conversion to ClC-3unc, markedly lowered ClC-4 protein levels largely abolishing compenstaion by ClC-4 in the KO, but not in KI mice. Hence, probably all vesicular CLCs depend on Cl-/H+ exchange activity for their bilogical function.
We recently discovered a new human channelopathy caused by mutations in CLCN6 (Polovitskaya et al., AJHG 2020). Whereas disruption of ClC-6 in mice leads to mild neuronal storage, a recurrent missense mutation that enhances ClC-6 currents and leads to a loss of it pH-dependence leads to very severe developmental delay and neurodegeneration. This point mutant leads to grossly enlarged lysosome-like vesciles in tranfected cells, an effcet that is abolished by converting ClC-6 into an uncoupled chloride conductor. Research Highlight
With the identification and characterizations of mutations in CLCN3 in several patients with neurodegenerative/developmental disorders we closed the last gap in human CLC channelopathies. As with CLCN7, we found that both loss and gain of function mutations in thias late endosomal Cl/H exchanger can cause severe pathology. Duncan et al., AJHG 2021
Disruption of the widely expressed plasma membrane Cl channel ClC-2 leads to various pathologies in mice and humans, including blindness, male infertility, and leukodystrophy. Surprisingly, our French collaborators very recently discovered a ClC-2 mutation in a patient with primary hyperaldosteronism, which is associated with high blood pressure. Together we analyzed the effect of this mutation, which affects a residue in a gating region we had identified back in 1992 (Gründer et al, Nature), in cell culture and frog oocytes. The mutation strongly increases ClC-2 currents, leading to a depolarizing Cl efflux in aldosterone-producing glomerulosa cells in the adrenal cortex. This depolarization leads to an increase in transcription of the enzyme aldosterone synthase (see Fernandes-Rosa et al., Nature Genet. 2018).
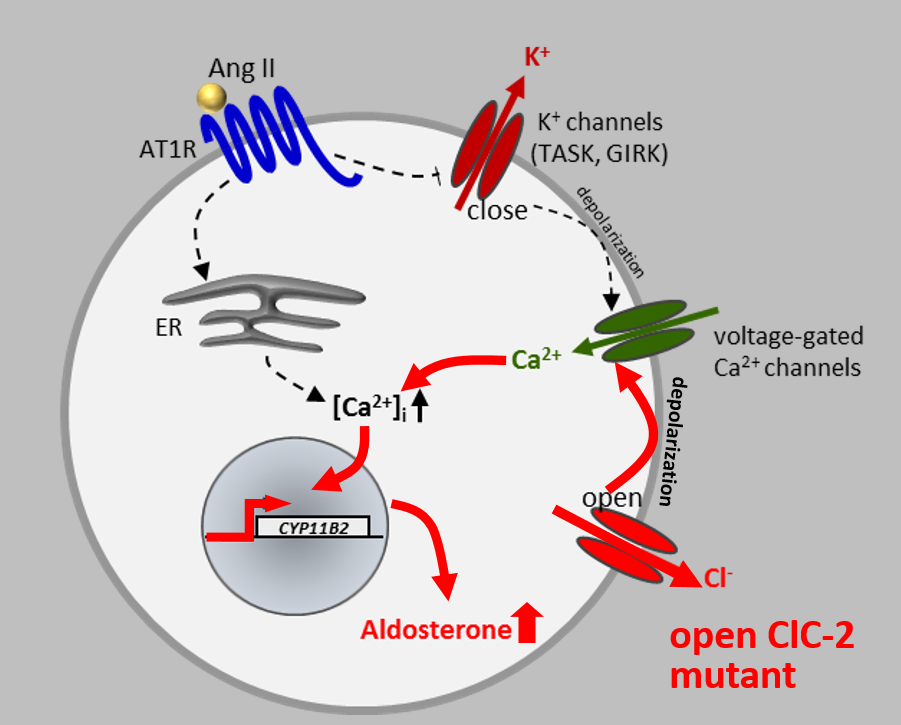
To prove that the opening of ClC-2 entails hyperaldosteronism, we generated a knock-in mouse model expressing a ClC-2 variant that carries a deletion in the N-terminal gating region. This deletion opens ClC-2 to the same degree as the human mutations we and others had identified in human patients. Patch-clamp analysis of adrenal glomerulosa cells showed that they were strongly depolarized by the dramatically larger chloride conductance. This resulted in increased aldosterone synthase transcription, high serum aldosterone, reduced serum renin, and arterial hypertension. These mice constitute the best mouse model for human aldosteronism to date. (see Göppner et al, Nature Comm. 2019).
We recently investigated cellular effects of ClC-2 disruption in additional mouse models, and continue to analyze the various disease-related roles of ClC-2 in more detail.
Das Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) gehört zum Forschungsverbund Berlin e.V. (FVB), einem Zusammenschluss von sieben natur-, lebens- und umweltwissenschaftlichen Instituten in Berlin. Die Einrichtungen sind Mitglieder der Leibniz-Gemeinschaft.
Leibniz-Forschungsinstitut für Molekulare Pharmakologie im Forschungsverbund Berlin e.V. (FMP)
Campus Berlin-Buch
Robert-Roessle-Str. 10
13125 Berlin, Deutschland




![[Translate to Deutsch:] background header](/fileadmin/_processed_/1/b/csm_Bacteriophage_final_v02_bb61a5ea72.jpg)

![[Translate to Deutsch:] link](/fileadmin/_processed_/0/6/csm_1_c223dcd589.jpg)

![[Translate to Deutsch:] background header](/fileadmin/_processed_/6/2/csm_forschergruppen_5_34300470f7.jpg)
![[Translate to Deutsch:] bachground picture](/fileadmin/_processed_/3/d/csm_Forschungsgruppen_ddee470de1.jpg)

![[Translate to Deutsch:] leibniz phd](/fileadmin/_processed_/3/f/csm_fmp-career-phd_aaf1698784.jpg)




![[Translate to Deutsch:] link](/fileadmin/_processed_/4/6/csm_Glaesernes_Labor-0032_3642a3cb69.jpg)


